Première cause de mortalité dans le monde, l’athérosclérose reste une pathologie souvent mal comprise. Pourtant, derrière l’infarctus du myocarde ou l’AVC se cache une maladie inflammatoire chronique complexe où le mécanisme de rupture de la plaque d’athérosclérose joue un rôle bien plus déterminant que la seule obstruction du vaisseau. Le groupe du Dr. Benoit Pourcet (UMR1011 Pr Bart Staels, Equipe 5 Dr Hélène Duez), maitre de conférence à l’université de Lille et chercheur à l’Institut Pasteur de Lille, décrypte pour nous les mécanismes biologiques de cette pathologie et expose les nouvelles stratégies thérapeutiques qui, au-delà du seul cholestérol, ciblent désormais l’inflammation pour mieux protéger les patients à haut risque cardiovasculaire.
Dr. Benoit Pourcet (UMR1011 Pr Bart Staels, Equipe 5 Dr Hélène Duez), maitre de conférence à l’Université de Lille et chercheur à l’Institut Pasteur de Lille

Sur quoi portent précisément vos recherches au laboratoire ?
Benoit Pourcet (BP) : Au laboratoire, nous cherchons à identifier des facteurs clés et des cibles pharmacologiques impliqués dans la régulation des mécanismes conduisant à l’infarctus du myocarde. On sait aujourd’hui que ce sont ces mécanismes qui sont les plus délétères. Plus précisément, c’est la rupture de la plaque d’athérosclérose qui provoque l’événement aigu. Pendant longtemps, on a pensé que la lente l’obstruction du vaisseau était l’événement le plus dangereux, mais c’est bien la rupture qui entraîne l’infarctus, l’accident vasculaire cérébral (AVC) ou l’artérite des membres inférieurs.
Pourquoi l’athérosclérose est-elle considérée comme une maladie chronique majeure de santé publique ?blématique majeure qui empêche la guérison complète du VIH chez les patients sous traitement ?
BP : C’est une excellente question car elle permet de redéfinir la nature même de cette pathologie. L’athérosclérose est avant tout une maladie inflammatoire chronique stérile de la paroi vasculaire, que l’on pourrait comparer, à cet égard, à la polyarthrite rhumatoïde ou à la goutte.
Le terme « chronicité » est ici fondamental : nos vaisseaux sont exposés de manière permanente à l’agent déclencheur de l’inflammation, qui est ici l’accumulation de cholestérol, et plus précisément des lipoprotéines de faible densité (LDL). Ces LDL servent à transporter le cholestérol du foie vers les tissus qui en ont besoin. Il faut d’ailleurs sortir de l’idée reçue du « mauvais cholestérol » car nous en avons un besoin vital pour nos membranes biologiques, nos hormones (sexuelles et glucocorticoïdes), la vitamine D ou les acides biliaires. C’est uniquement l’excès et l’accumulation prolongée qui déclenchent la formation de la plaque.
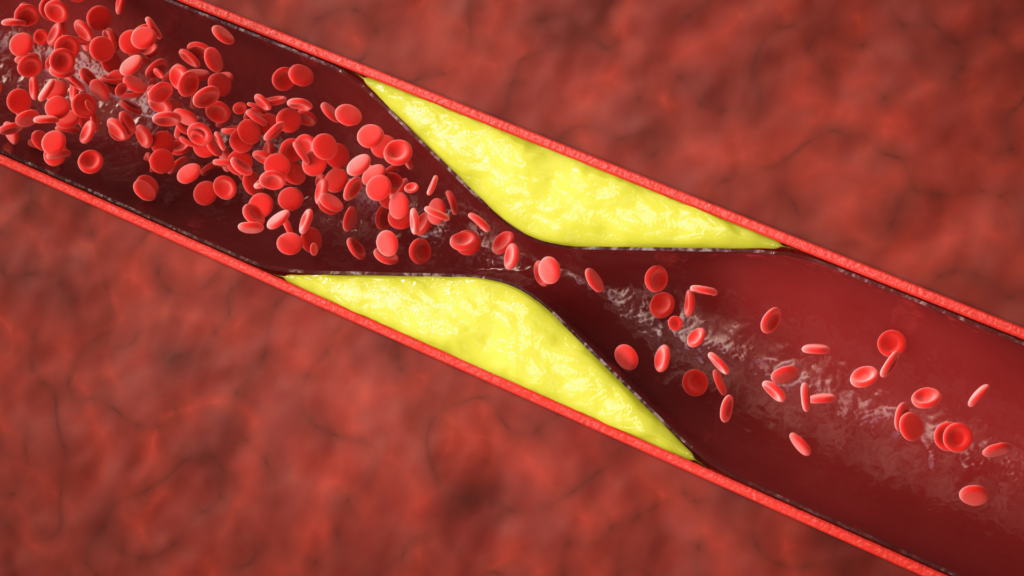
À partir de quel moment cette pathologie commence-t-elle à devenir préoccupante ?
Benoit Pourcet : C’est un processus très long. On commence généralement à former des plaques dès l’âge de 4 ans. La plaque se complexifie ensuite sur des décennies, jusqu’à la rupture qui peut survenir, selon les facteurs de risque, entre 40 et 90 ans.
C’est un enjeu de santé publique massif car, à l’échelle de la planète, les maladies cardiovasculaires restent la première cause de mortalité. Si le cancer est passé en tête dans certains pays occidentaux, les maladies cardiovasculaires explosent en Inde et en Chine en raison des changements d’habitudes alimentaires. Les pathologies les plus meurtrières, à savoir les maladies ischémiques coronariennes et certains AVC, sont majoritairement la conséquence directe de la formation de ces plaques d’athérome.
Que se passe-t-il concrètement dans les artères lors du développement de cette maladie ?
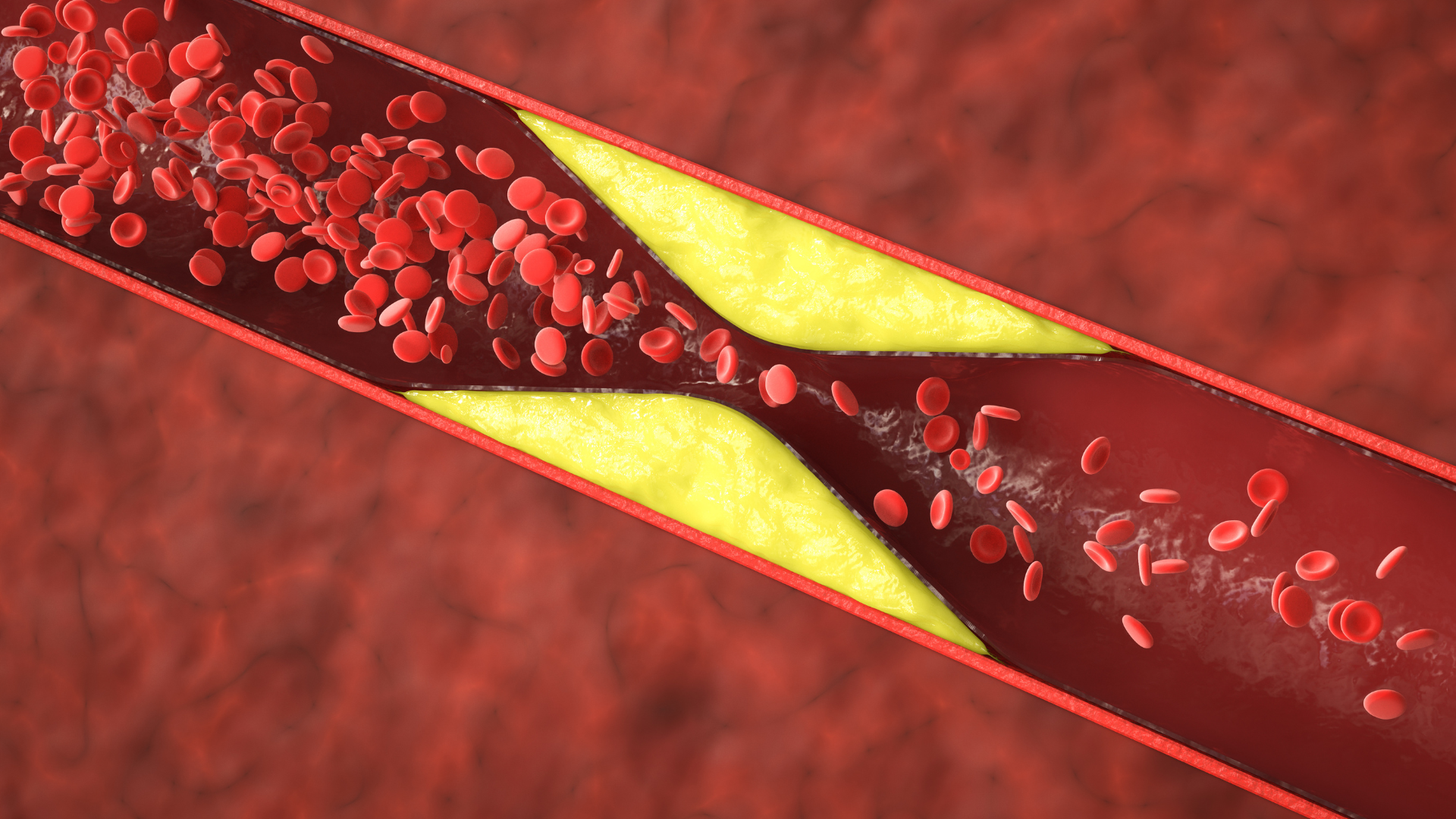
BP : C’est un point sur lequel j’insiste beaucoup, car il existe une idée reçue très tenace. Pour illustrer, on imagine souvent que l’athérosclérose ressemble à des impuretés qui viennent boucher une canalisation en se collant à la paroi. En réalité, ce n’est pas du tout cela : les lipides s’insinuent à l’intérieur même de la paroi du vaisseau. Le vaisseau est composé de trois couches, et le cholestérol pénètre dans cet espace, ce qui déclenche une véritable alerte pour votre système immunitaire.
Comment le corps réagit-il à cette intrusion de lipides ?
BP : Comme pour n’importe quel corps étranger, qu’il s’agisse d’un virus ou d’une bactérie, l’organisme déclenche une réponse inflammatoire. Des cellules immunitaires appelées macrophages arrivent sur place pour « manger » (phagocyter) ce cholestérol. Au début, c’est un mécanisme protecteur : les macrophages tentent de renvoyer ce cholestérol vers le foie pour qu’il soit éliminé, via ce qu’on appelle les lipoprotéines de haute densité (HDL) et que l’on associe souvent maladroitement à ce que l’on appelle « bon cholestérol ».
Le problème devient dangereux à cause du côté chronique de l’exposition. À force de se nourrir de cholestérol, les macrophages finissent par mourir, d’abord par apoptose, puis par nécrose. En mourant, ils déversent tout leur contenu lipidique dans la paroi, formant ce que nous appelons un cœur nécrotique. C’est cette accumulation de débris et de gras qui devient extrêmement toxique pour la plaque.
La plaque peut-elle tout de même se stabiliser ?
BP : Oui, le corps essaie de se défendre. Pour contenir ce cœur nécrotique, des cellules de la paroi vont injecter du collagène afin de créer une chape fibreuse pour stabiliser l’ensemble. J’utilise souvent l’analogie du béton armé : le collagène joue le rôle des fils de fer qui viennent renforcer la structure. Tant que cette chape est épaisse, la plaque est stable et le patient ne risque pas grand-chose dans l’immédiat.
Qu’est-ce qui provoque alors l’accident aigu, comme l’infarctus ?
BP : Tout bascule lorsque l’inflammation devient trop forte. Les macrophages présents peuvent alors se mettre à « digérer » la plaque. D’autres phénomènes, comme la calcification vasculaire ou la formation de petits vaisseaux dans les plaques (néovascularisation), vont venir affiner cette chape fibreuse.
Plus la chape s’affine, plus la plaque devient instable et sujette à la rupture. Le moment critique arrive lorsque la chape rompt : les fibres de collagène internes sont subitement exposées aux facteurs de coagulation du sang. Cela déclenche instantanément la formation d’un caillot qui vient boucher totalement l’artère, stoppant l’irrigation du muscle cardiaque ou du cerveau. C’est ainsi que survient l’infarctus.
Quelle est la différence entre la progression lente de la maladie et l’événement aigu de la rupture ?
BP : Il faut bien distinguer deux scénarios dans l’évolution de la plaque d’athérome. Le premier est celui d’une progression lente où l’épaississement de la plaque rétrécit progressivement la « lumière » du vaisseau, ce qui n’entraîne aucun symptôme au repos mais provoque une ischémie d’effort dès que les besoins en oxygène des organes augmentent. C’est ce qui se passe dans l’angine de poitrine (angor) lorsque le patient ressent une vive douleur lors d’un effort même peu intense comme la marche par exemple, mais celle-ci disparaît dès qu’il se rassoit car la demande en oxygène diminue et le vaisseau rétréci suffit de nouveau à la tâche. Ce type d’événement doit d’ailleurs être un signal absolu d’alerte pour aller consulter son médecin. À l’opposé, l’événement aigu relève d’une rupture brutale ou d’une érosion de la plaque, qui déclenche la formation instantanée d’un caillot et une obstruction totale du vaisseau. Dans ce cas, le tissu n’est plus du tout irrigué, il ne reçoit plus de nutriments ni d’oxygène, on dit qu’il est ischémié, c’est ce que l’on appelle l’infarctus ; la douleur ne passe pas avec le repos et nécessite une prise en charge immédiate pour limiter l’étendue des lésions et garantir une meilleure récupération.
Qu’est-ce qui a orienté votre parcours vers l’étude de ces pathologies ?
BP : Mon parcours a débuté par une classe préparatoire à Poitiers, suivie d’études de biochimie à Bordeaux, où je me suis passionné très tôt pour la signalisation cellulaire. Ces mécanismes de transduction du signal m’intéressaient particulièrement car ils constituent un réservoir immense de cibles pharmacologiques dont la dérégulation conduit à de nombreuses pathologies. Au départ, je me destinais plutôt à la recherche sur le cancer, mais l’opportunité d’une thèse à l’Institut Pasteur de Lille, dans l’équipe de Bart Staels, a modifié ma trajectoire. J’y ai travaillé de manière très moléculaire sur PPAR-alpha, une cible des fibrates liée au contrôle du métabolisme lipidique qui avait été découverte ici même.
C’est véritablement lors de mon post-doctorat à l’University College of London, dans l’équipe d’Inés Pineda Torra, que j’ai plongé dans le domaine de l’athérosclérose. Une fois confronté au sujet à Londres, j’en suis tombé éperdument amoureux. Ce qui me passionne, c’est justement cette complexité multidisciplinaire : l’athérosclérose est au carrefour de l’inflammation, du métabolisme, du remaniement des tissus et des enjeux épidémiologiques comme le diabète. Aujourd’hui, nos recherches se concentrent sur une « niche » essentielle : comprendre les mécanismes qui induisent l’entrée des lipides dans la paroi et ceux qui conduisent à la rupture de la plaque, afin de développer des traitements pour les patients à très haut risque cardiovasculaire.
Quelles sont les perspectives thérapeutiques pour demain ?
BP : Jusqu’à présent, en dehors des règles hygiéno-diététiques qui sont fondamentales, la prise en charge reposait quasi exclusivement sur les traitements hypocholestérolémiants, mais la communauté scientifique et médicale s’accorde désormais sur la nécessité de mettre en place des thérapies combinatoires intégrant une dimension anti-inflammatoire, particulièrement pour les patients à très haut risque cardiovasculaire comme les patients souffrant de diabète. On préconise maintenant l’usage d’anti-inflammatoires non spécifiques comme la colchicine, traditionnellement utilisée pour la goutte, tandis que des recherches de pointe, notamment celles de Ziad Mallat à Cambridge ou de Novo Nordisk, valident l’intérêt de cibler des interleukines comme l’IL-2 ou l’IL-6, dont les traitements sont actuellement en phases finales de développement. Au sein de notre équipe, nous avons d’ailleurs identifié une cible pharmacologique prometteuse qui permettrait d’agir de manière globale et proposer une meilleure prise en charge en agissant simultanément sur les lipides, l’inflammation et les mécanismes critiques de rupture de la plaque.